PK Explorer Tutorial

This tutorial demonstrates how PK Explorer can be used for exploring the effect of lipophilicity on absorption.
Prior to following this tutorial, make sure that you are in Expert mode, and in the Expert Panel set GALAS versions of all available algorithms as defaults.
The drug-like compound shown on the right was selected for the demonstration.
Enter the structure of this compound by pasting it from the clipboard (
 ), by opening the molecular file (
), by opening the molecular file ( ), by drawing it in the built-in editor
), by drawing it in the built-in editor  or by entering the following SMILES notation (
or by entering the following SMILES notation ( ): OC1C(O)C(=O)C(OC1CO)C3=CC(=O)c2c(O)cccc2C3=O
): OC1C(O)C(=O)C(OC1CO)C3=CC(=O)c2c(O)cccc2C3=OIf tautomer selection dialog box pops up, simply press OK without changing the default tautomer.
PK Explorer will simulate absorption, disposition and elimination of the test compound at default dose of 50 mg automatically.
The tested compound has poor bioavailability, which is predicted to be partly due to extensive first-pass effect, partly due to poor absorption. We will ignore the first-pass effect, as in this example we would like to explore the effect of logP on absorption.
The predicted bioavailability of tested compound even in the absence of presystemic metabolism is incomplete (<50%), and bioavailability also depends on logP.
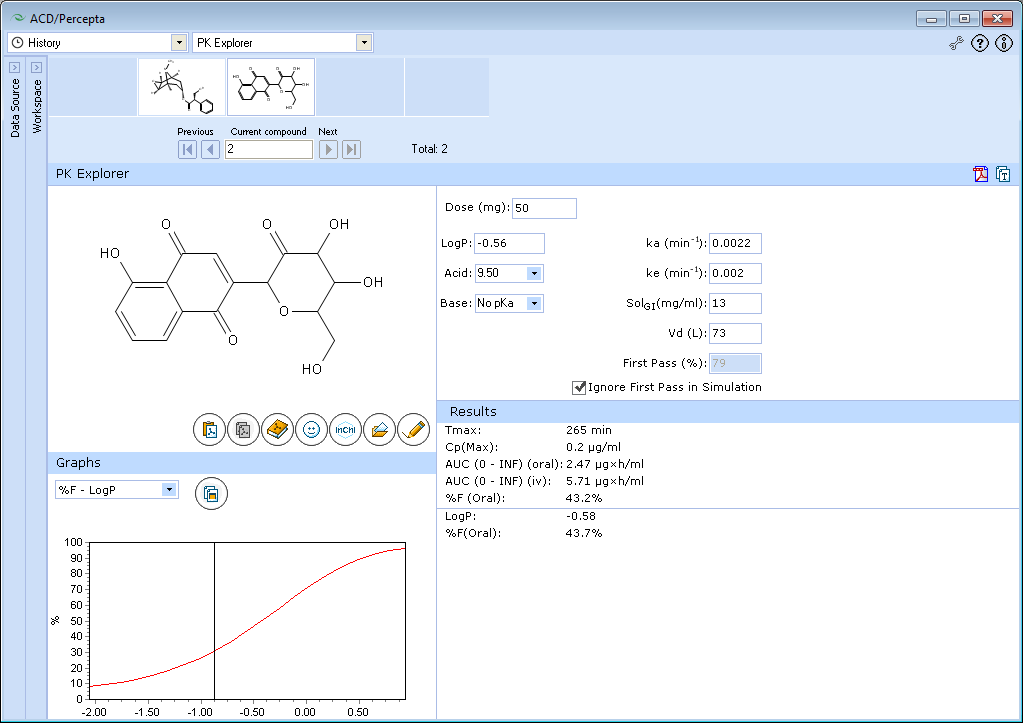
The compound is hydrophilic and its absorption is limited by poor permeability. If this compound's logP would be lower by further 0.5 units, the extent of absorption would be less than 30%. Absorption can be improved by addition of hydrophobic groups. In this case, the optimal logP for absorption would be in the range 0.5-1.5. The predicted extent of absorption in this logP range exceeds 90%.
Explore the influence of logP on oral absorption:- a. Set the slider at logP = 0.88
- b. See the exact value of %F oral in the lower part of the Results Pane
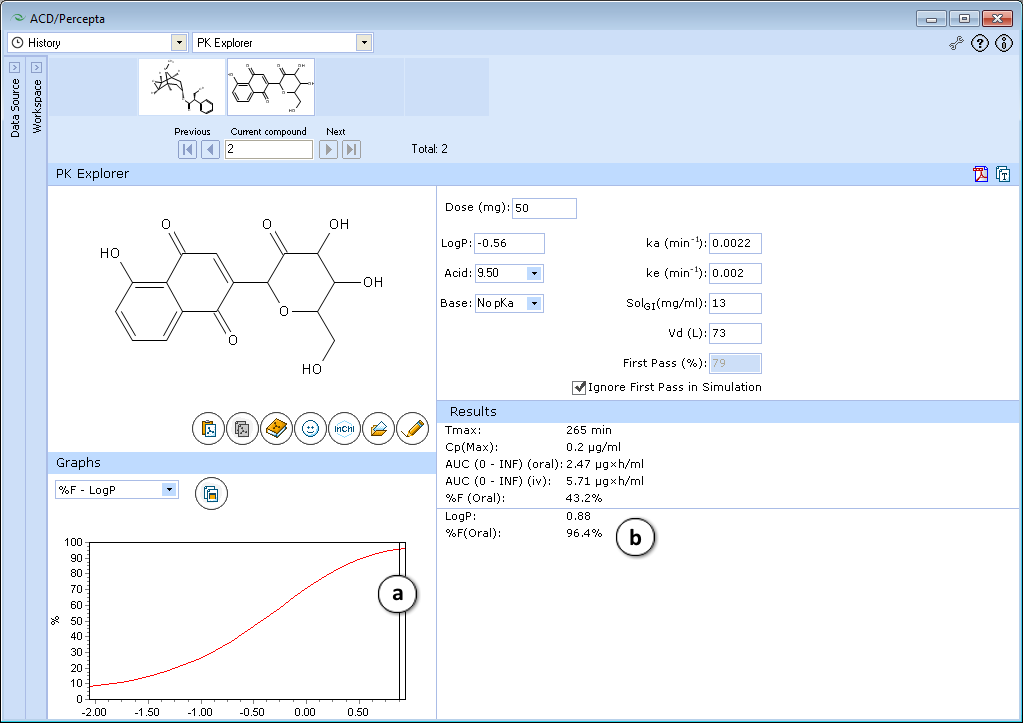
Eterification of phenolic group significantly improves absorption both by increasing hydrophobicity and by blocking the weak acidic group.
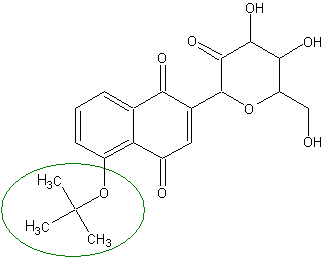
Enter the structure of the modified compound by pasting it from the clipboard (
), by opening the molecular file (), by drawing the tert-butyl group in the built-in editor or by entering the following SMILES notation (): OC1C(O)C(=O)C(OC1CO)C3=CC(=O)c2c(OC(C)(C)C)cccc2C3=OThe extent of absorption of the modified compound is estimated to be greater than 95%.

Let’s assume that the drug will be used at 200 mg, and experimental logP of previously modified test compound is 1.0 log unit greater than predicted.

Perform the corresponding changes in PK Explorer window and perform the simulation with altered input values (lines indicate the parameters that were affected by changes of logP):
- The Undo button indicates that value of parameter was changed. Clicking on this button restores the initial calculated value. Predicted pharmacokinetic parameters (elimination rate - ke, volume of distribution - Vd, etc.) can also be replaced by clinical data if available. Solubility of the compound in gastrointestinal tract (SolGI) can be altered too.
- Click to perform a new simulation using the currently specified parameter values.
- Lines indicate that ka and SolGI are automatically recalculated according to new logP values.
- Select the %F-Dose graph.
- Set the slider at mg = 200 by clicking and dragging it.
The predicted absorption with these new parameters is still good, but it becomes strongly dose-dependent. This pattern is undesirable as dose-dependent absorption is usually highly variable.
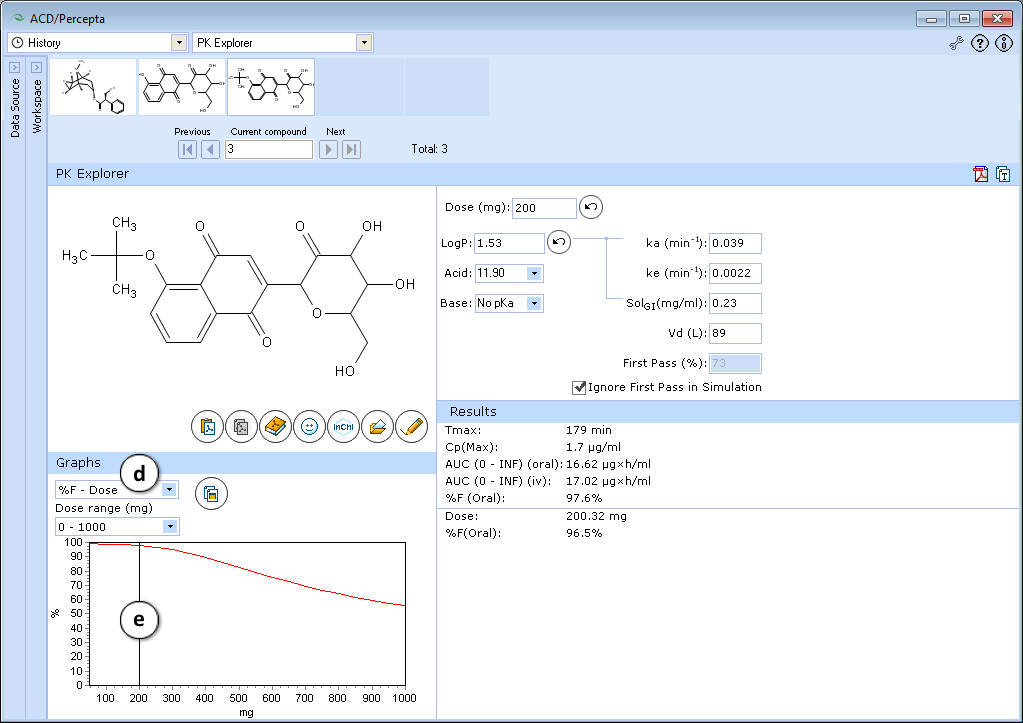






Conclusion: The absorption of the test compound can be improved with small changes in logP (0.5-1 log unit). Further increase in hydrophobicity (e.g. by blocking other alcohol groups) is undesirable.