In this tutorial we will walk through a real-world scenario, demonstrating how to use ACD/Structure Designer to facilitate lead optimization process. This example will follow the same route that had been actually undertaken during the discovery of cardiotonic drug sulmazole (Kutter E, Austel V. Arzneim.-Forsch.1981, 31, 135.)
In this case, the initial lead compound had CNS side effect, which manifested as “bright flashes” seen by some patients treated with this compound, while the final lead - sulmazole - showed the desired cardiotonic activity without any CNS-related side effects. Therefore, the objective of our in silico lead optimization process is reducing brain penetration of the target compound in order to avoid CNS activity.
For the purpose of this tutorial, all physicochemical property prediction algorithms (LogP, pKa, Solubility) should be set to the GALAS version (configurable via the Expert Panel ).
Input the structure of the initial lead: load any Prediction Module, click button, and paste the following SMILES string: c1(nc2ncccc2[nH]1)c1c(cc(OC)cc1)OC
If the Select tautomeric form dialog box pops up, leave the default form selected and simply press OK.
Switch to Structure Designer workspace. Note the output of ADME Profile/Project Objectives window.
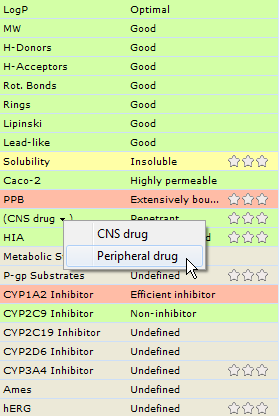
The compound is predicted CNS Penetrant, however, this property is colored green indicating its favorable contribution to ADME profile, since the option for optimizing CNS drugs is on by default. Select the Peripheral drug entry from the arrow menu to reverse the target BBB penetration score. CNS (PS drug) field should now be colored red.
Also note the predictions of other ADME properties. The only other property in the unfavorable range is PPB, due to extensive plasma protein binding. However, this does not necessarily need too be improved, as it also contributes to restricting the molecule from brain.
Set the highest priority to CNS field by marking all three stars in the Importance column.
In the right-hand side, the property scales are now filled with color gradients, and the property ranges enclosed by sliders all fit in the unfavorable orange-red zone.
Pre-filter the list of substituents. In this example we are using a built-in substituent set included in ACD/Structure Designer, and we will filter it to display only acyclic substituents in order to preserve the overall shape of the molecule.
Select Substituents > Built-in substituents data source.
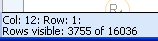
Select the newly calculated column Number of Rings, and set up the filter in the Column Menu, so that only substituents with zero rings are shown.
The status bar now shows that 3755 of 16036 rows fulfill the filter criteria:
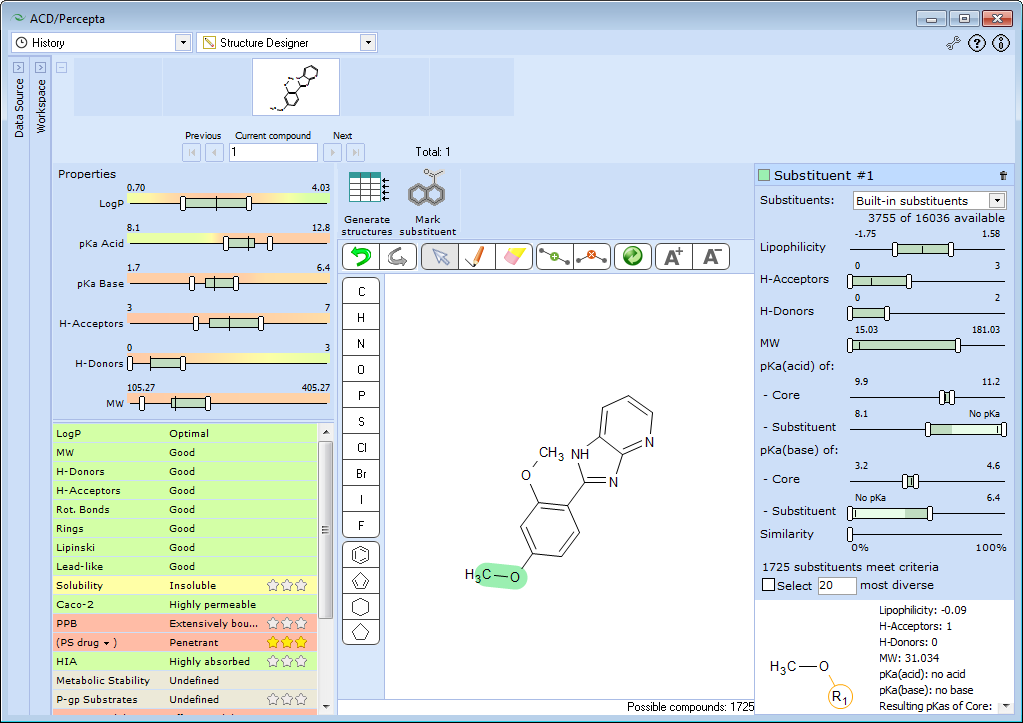
Switch back to History data source and Structure Designer workspace, and mark the substituent you wish to optimize. In the Structure Pane, select Arrow tool, lasso around the –OCH3 group in para-position and press the Mark substituent button. The panel Substituent #1 and the number of suitable substituents found in the database appears.
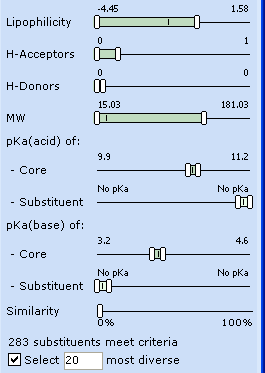
Obviously, the compound enters the brain since it is lipophilic enough. To generate less lipophilic analogs, push the left LogP slider in the ADME Profile/Project Objectives window all the way to the left, down to LogP = -2.00.
This panel is linked with the Substituent Panel and the changes are automatically reflected therein, with the left Lipophilicity slider set the lowest available value.
Explore other properties in the Substituent Panel. The methoxy- group in the initial lead contains one H-bond acceptor and no H-bond donors. To ensure that essential target interactions are not disrupted in the analogs, restrict the substituents to those that do not introduce additional H-acceptor and H-donor sites.
The lead compound does not contain any ionizable groups. To preserve neutral ionization state, push both pKa(acid) of substituent and pKa(base) of substituent sliders towards "No pKa" zone, so that only neutral fragments are displayed.
Mark “Select 20 most diverse” checkbox to reduce the number of generated analogs.
Click the Generate structures button. If a "Read-only data source" message appears, click Yes to create a new project that will store the newly generated structures.
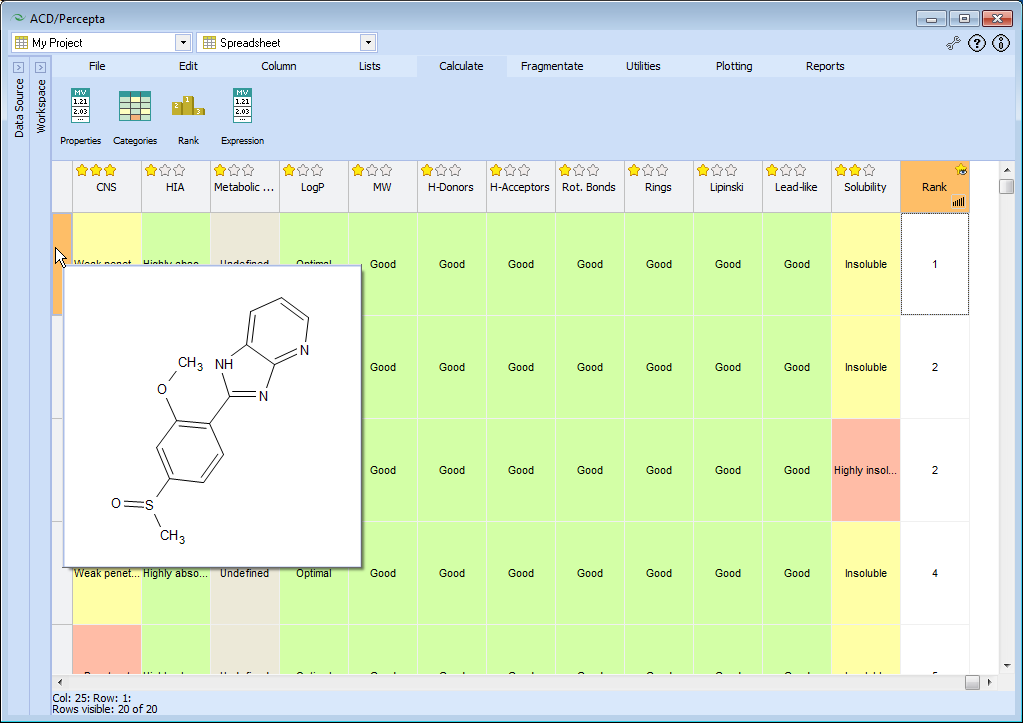
To analyze the analogs suggested by the software, select My Project in the Data Source Panel, and switch to Spreadsheet in the Workspace Panel.
Predict the full ADME/Tox profile of the new molecules by pressing Calculate > Categories button, and selecting all categories available for calculation.
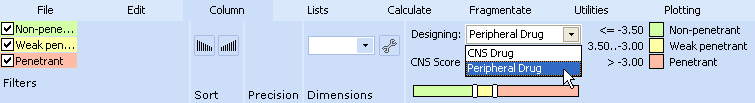
Wait until the calculation finishes, then navigate to CNS column and set Designing > Peripheral drug in the Column menu.
Rank the analogs by their ADME/Tox profiles: click Calculate > Rank button, and select all available categories.
Press button in the Rank column and select three stars for CNS category, as we have done before in the Structure Designer workspace.
Sort the analogs by their ranks. The molecule that obtains Rank 1 is the target compound sulmazole.
These results illustrate, how ACD/Structure Designer can help in finding lead compounds with better ADME profiles with respect to the defined project objectives.

 button, and paste the following SMILES string: c1(nc2ncccc2[nH]1)c1c(cc(OC)cc1)OC
button, and paste the following SMILES string: c1(nc2ncccc2[nH]1)c1c(cc(OC)cc1)OC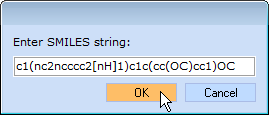




 button in the Rank column and select three stars for CNS category, as we have done before in the Structure Designer workspace.
button in the Rank column and select three stars for CNS category, as we have done before in the Structure Designer workspace.